It should now be clear that population size will affect the number of alleles present in a population. But small population sizes also introduce a random element called genetic drift into the population genetics of organisms.
Genetic drift is a process in which allele frequencies within a population change by chance alone as a result of sampling error from generation to generation. Genetic drift is a random process that can lead to large changes in populations over a short period of time. Random drift is caused by recurring small population sizes, severe reductions in population size called "bottlenecks" and founder events where a new population starts from a small number of individuals. Genetic drift leads to fixation of alleles or genotypes in populations. Drift increases the inbreeding coefficient and increases homozygosity as a result of removing alleles. Drift is probably common in populations that undergo regular cycles of extinction and recolonization. This may be especially important in natural ecosystems where both plants and pathogens are likely to have a patchy distribution where each patch is a small population.
Because allele frequencies do not change in any predetermined direction in this process, we also call genetic drift "random drift" or "random genetic drift." The sampling error can occur in at least three ways. We will consider these in the context of pathogen populations in plant pathosystems:
- Small recurring population size occurs when there are not many host plants in the area to infect, or when the environment is not optimal for infection.
- A genetic bottleneck, or severe reduction in population size, occurs when the plant population is removed (e.g. harvest of the crop), or when the environment changes to prevent infection of the plant or to kill the pathogen directly (e.g. periods of hot, dry weather or a deep freeze).
- A founder effect occurs when a small number of individuals, representing only a small fraction of the total genetic variation in a species, starts a new population. A founder event occurs when one or two infected plants slip through a quarantine and introduce a disease into an area where the disease did not previously exist.
The magnitude of genetic drift depends on Ne, the effective population size, for the population. Ne is rarely the actual number of individuals in the population (also called N or the census size). Ne is a theoretical number that represents the number of genetically distinct individuals that contribute gametes to the next generation. Ne can also be thought of as the number of genetically distinct interbreeding individuals in a population. Ne is not easy to quantify because it is affected by reproduction and breeding strategies (inbreeding, outcrossing, asexual reproduction), and is dependent on the geographical area over which a population is sampled. Ne is not easy to define for fungal pathogens that undergo a mixture of sexual and asexual reproduction because the absolute number of individuals can be very large while the number of different genotypes that sexually recombine can be relatively small. An analysis of field populations of the wheat pathogen Mycosphaerella graminicola indicated Ne of at least 70 strains per square meter (Zhan et al., 2001).
We can calculate how much genetic drift we expect to find in a population if we know the effective population size. The expected variance in the frequency of an allele (call this frequency p) subject to genetic drift is:
Var (p) = 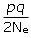 after one generation of genetic drift for diploid organisms.
after one generation of genetic drift for diploid organisms.
After many generations of genetic drift, an equilibrium will be reached. At equilibrium we expect that:
Var (p) = p0q0
Where p0 and q0 are the initial frequencies of the two alleles at a locus.
If p0=q0=0.5 and Ne = 50 then Var (p) = 0.0025
The standard deviation of (p) = (0.0025)0.5 = 0.05.
The standard deviation is the average absolute value of the expected difference among populations after one generation of drift and is approximately equal to the expected change in allele frequency ( p) within each population. Thus in a population of 50 individuals, with two alleles beginning at equal frequencies, we expect the allele frequencies to change by about 5% each generation.
p) within each population. Thus in a population of 50 individuals, with two alleles beginning at equal frequencies, we expect the allele frequencies to change by about 5% each generation.
The degree of change increases as the population size decreases.
If p0=q0=0.5 and Ne = 5 then Var (p) = 0.05
The standard deviation of (p) = (0.05)0.5 = 0.22
In this case, a population that has only five individuals is expected to experience random changes in allele frequencies of about 22% each generation.
The chance of fixing an allele due to genetic drift depends on the effective population size as well as the frequency distribution of alleles at a locus. To "fix" an allele means that the allele is present at a frequency of 1.0, so all individuals in the population have the same allele at a locus. Large effective population sizes and an even distribution in allele frequencies tend to decrease the probability that an allele will become fixed (Figure 5). Alleles that occur at a low frequency are usually at a disadvantage in the process of genetic drift. Low-frequency alleles face a higher probability of disappearing from a population than alleles that occur at a higher frequency. Under a scenario of pure genetic drift, the probability of fixation of an allele in a population is its initial frequency in the population. If the initial frequency of an allele is 0.01, then there is a 1% chance that this allele will be fixed in this population. Thinking in a different way, if the initial allele frequency was 0.01 in 100 different populations, then we expect that approximately 1 of those populations would become fixed for this allele after many generations of random genetic drift.
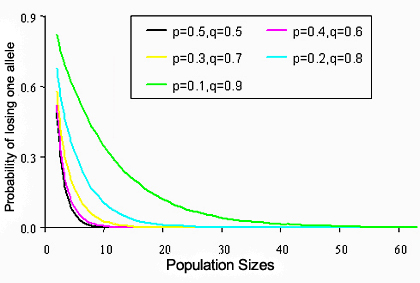
Figure 5. The probability that an allele will drift away in any single generation in a two-allele model with different initial frequencies and different effective population sizes.
The consequences of genetic drift are numerous. It leads to random changes in allele frequencies. Drift causes fixation of alleles through the loss of alleles or genotypes. Drift can lead to the fixation or loss of entire genotypes in clonal (asexual) organisms. Drift leads to an increase in homozygosity for diploid organisms and causes an increase in the inbreeding coefficient. Drift increases the amount of genetic differentiation among populations if no gene flow occurs among them.
Genetic drift also has two significant longer-term evolutionary consequences. Genetic drift can facilitate speciation (creation of a new species) by allowing the accumulation of non-adaptive mutations that can facilitate population subdivision. Drift also facilitates the movement of a population from a lower fitness plateau to a higher fitness plateau according to the shifting balance theory of Sewall Wright.
The amount of population subdivision is expected to increase because of the random losses of alleles that occur in different populations. In addition, random changes in allele frequencies are expected to occur in different populations, and these random changes tend to make populations become differentiated. Finally, small effective population sizes increase the likelihood that mating events will occur between close relatives, leading to an increase in inbreeding and subsequent loss of heterozygosity.
In agroecosystems, pathogen populations usually become very large as a result of the genetic uniformity of the host plant, so genetic drift may not play a large role in the evolutionary process within a farmer's field in the real world. Few experiments have been conducted to test this hypothesis. But there is lots of evidence for founder effects in agroecosystems, especially in Australia, because it was the continent most recently colonized by Europeans who introduced first their crops and then their crop diseases. In natural ecosystems, genetic drift may play a more prominent role in the evolution of pathogens because host populations are genetically diverse and have a patchy distribution, so pathogen population sizes are not so large, and bottlenecks probably occur frequently in these natural populations. We will return to this theme after introducing the concept of metapopulations.
Mycosphaerella graminicola causes Septoria tritici leaf blotch on wheat. McDonald and colleagues used restriction fragment length polymorphism (RFLP) markers to determine the genetic structure of this pathogen worldwide and found that all populations collected from different geographic locations had similar frequencies of common alleles except the populations collected from Australia and Mexico (Zhan et al., 2003). The Australian and Mexican populations had significantly lower gene diversity (shown in Table 1), fewer alleles at each locus, fixed alleles at several RFLP loci, and the gene frequencies were significantly different from populations at other locations. In Australia, this is probably due to a founder effect whereby only a relatively small number of individuals arrived on this continent with the introduction of modern agriculture. The Patzcuaro, Mexico population was sampled from a breeding nursery used by CIMMYT to screen for resistance to this pathogen. This nursery is located far away from wheat production areas (hence, it has a limited potential for influx of natural inoculum) and was inoculated with a limited number of strains, presenting a clear example of genetic drift due to a small founding population and continued geographical isolation. In contrast, the Israeli population had the highest level of gene diversity, consistent with the hypothesis that the Middle East is the center of origin for this pathogen.
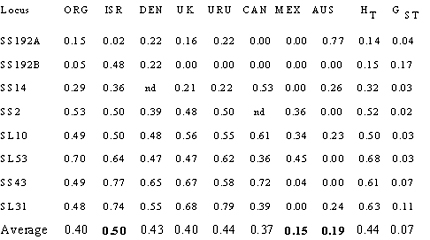
Table 1. The effect of genetic drift on gene diversity at RFLP loci in Mycosphaerella graminicola populations from Oregon, Israel, Denmark, United Kingdom, Uruguay, Canada, Mexico, and Australia. Populations from Mexico and Australia show low gene diversity consistent with founder effects while the Israeli population shows highest gene diversity consistent with the center of origin.
An extreme example of genetic drift due to a bottleneck is the population of the Phytophthora infestans pathogen that causes late blight of potatoes. It appears that the original global pandemic was caused by a single clone that escaped out of Mexico and into North America, was introduced into Europe (causing the Irish potato famine) and then was transported around the world as a result of human commerce (Goodwin et al. 1994).
Stripe rust of wheat (Puccina striiformis) in Australia shows evidence for a single founder event. P. striiformis was introduced into Australia in 1979. Only one race was found in 1979-1980, corresponding to a race found in Europe, suggesting that Europe was the source of the introduction. Since the original introduction, mutations have created new pathotypes in the single introduced genetic background (Steele et al. 2001).
Chestnut blight (Cryphonectria parasitica) in North America also shows some characteristics of a founder population as it has much less genetic diversity than populations in Asia. It appears that the center of diversity and possible center of origin is in Japan (Milgroom et al. 1996).
Go to Knowledge Test for Interactions/Genetic Structure
Go to References
Next Section